
-
摘要:目的 探讨肺移植治疗囊性纤维化(CF)的疗效和预后。方法 回顾性分析行同种异体双肺移植术治疗1例终末期CF受者的临床资料,对CF受者的临床特点、诊断方法及治疗策略进行总结。结果 本例受者幼年发病,反复咳嗽、咳浓痰30年,肺部反复感染合并急性加重、慢性鼻窦炎及极重度营养不良,肺移植术前因呼吸肌无力难以脱离有创呼吸机,长期入住重症监护室(ICU)。影像学检查提示双肺多发囊柱状支气管扩张伴感染,经汗液测试和基因检测进一步确诊为CF。该受者2017年8月17日接受双肺移植术后经过康复治疗,肺功能逐渐恢复正常,康复后至投稿日已2年余,享有与同龄健康人同等的生活质量。结论 肺移植术作为终末期CF的有效治疗手段,不仅能挽救患者生命,而且还能显著提高患者的生活质量。Abstract:Objective To investigate the clinical efficacy and prognosis of lung transplantation in the treatment of cystic fibrosis (CF).Methods Clinical data of one patient with end-stage CF undergoing allogeneic bilateral lung transplantation were retrospectively analyzed. Clinical characteristics, diagnostic methods and treatment strategies of the CF recipient were summarized.Results The recipient had suffered from relevant symptoms since childhood including repeated cough and purulent sputum for 30 years, complicated with recurrent pulmonary infection combined with acute exacerbation, chronic sinusitis and extremely severe malnutrition. Prior to lung transplantation, the patient had to depend upon the invasive ventilator due to respiratory muscle weakness, and admitted to intensive care unit (ICU) for a long time. Imaging examination revealed multiple cystic columnar bronchiectasis accompanied with infection in bilateral lungs. The diagnosis of CF was further confirmed by sweat test and gene detection. The recipient underwent bilateral lung transplantation on August 17, 2017 and received rehabilitation treatment. The lung function was gradually restored to normal. The recipient had obtained the same quality of life to the healthy counterparts since the date of manuscript submission (over 2 years).Conclusions Lung transplantation is an efficacious treatment for end-stage CF, which can not only save patients' lives, but also significantly improve the quality of life of patients.
-
囊性纤维化(cystic fibrosis,CF)是一种常染色体隐性遗传的多系统疾病,该疾病由编码囊性纤维化跨膜电导调节因子(cystic fibrosis transmembrane conductance regulator, CFTR)蛋白的基因突变引起[1]。CF最常见于北欧白人[2],过去认为CF在亚洲人群中属于罕见病,但近年来随着对该疾病的认识加深及诊疗手段的提高,研究发现CF在中国人中发病率明显升高。本文报道1例32岁CF受者,通过对受者的临床特点、诊断手段及最终接受肺移植手术的治疗经过进行回顾,并检索CF诊治的相关文献,以期提高临床对这类疾病的认识及治疗水平。
CF主要表现为幼年即出现反复呼吸道感染、支气管扩张、慢性并进行性加重的阻塞性肺疾病,反复腹泻、脂肪泻、营养不良,合并肝硬化,伴汗液Na+、Cl-异常增高,男性不育等[3]。在过去的60年中,CF患者中位生存期较以前延长,在发达国家其中位生存期已超过40岁[1]。尽管近年来对CF的治疗方法有很多改善,生存率已经得到提高,但进行性加重的呼吸衰竭依旧是CF死亡的主要原因。目前,肺移植仍是终末期CF患者的重要治疗手段[4]。
1. 临床资料
受者,女,32岁。因“反复咳嗽、咳脓痰30年,咯血20年,加重4年”于2015年6月收入广州医科大学第一附属医院呼吸内科。受者幼年起病,反复咳嗽、咳痰,伴痰中带血,反复多次住院治疗。近年来每次感冒后出现咳嗽、咳痰反复加重,咳大量黄脓痰,伴反复咯血,气促进行性加重,外院诊断为“双侧支气管扩张合并感染、慢性多鼻窦炎、Ⅱ型呼吸衰竭、重度营养不良”,予以内科治疗效果欠佳,呼吸衰竭进行性加重,活动耐量持续下降,需要24 h持续氧疗,由于无法脱离氧疗只能床边活动, 后病情呈慢性、进行性加重,需要长期卧床、24 h依赖无创呼吸机。求诊于我院前,受者因反复Ⅱ型呼吸衰竭、肺性脑病多次入住重症监护室(intensive care unit,ICU), 后因严重营养不良、呼吸肌无力难以脱离有创呼吸机,长期入住我院ICU。
受者就诊我院时已属于终末期肺疾病,无法完成肺功能检查,幼年时外院肺功能检查提示:极重度混合性通气功能障碍。移植前多次深部痰培养结果提示:多重耐药或泛耐药鲍曼不动复合菌及铜绿假单胞菌阳性。胸部CT示:双肺广泛囊柱状支气管扩张,支气管管壁增厚,双肺散在黏液栓及树芽征。鼻窦CT平扫:双侧上颌窦、筛窦、右侧蝶窦炎症。心脏彩色多普勒超声示:射血分数(ejection fraction,EF)为73%,三尖瓣反流(轻度),肺动脉高压(轻度),左室收缩功能测值未见异常。受者入院后,结合其幼年起病的病史及临床表现,考虑CF可能性大,进行了2次汗液测试,Cl-浓度分别为112 mmol/L及82 mmol/L(正常参考值< 60 mmol/L),经基因检测结果明确诊断为CF。
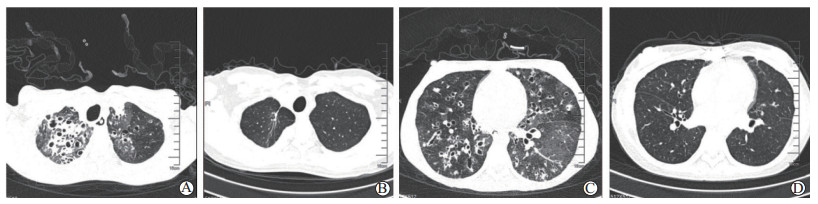
针对该患者,于2017年8月17日行同种异体双肺移植术,术中体外膜肺氧合(extracorporeal membrane oxygenation,ECMO)辅助,手术时间10 h,术中出血量为13 750 mL,出血量多于普通患者,主要原因为长期严重缺氧,胸腔内血管侧枝循环丰富。术后即撤除ECMO,术后11 d拔除气管插管。术后12 d转入移植病区,后再次因肺部感染及颅内真菌感染转入ICU继续治疗。该受者术后康复困难,康复时间为162 d,显著长于其他肺疾病受者移植术后的平均康复时间。影响受者康复的主要原因为严重肌肉无力、营养不良、反复加重的感染等综合因素。出院后定期门诊随诊,肺功能逐渐改善,活动耐量如同正常人。术后2年胸部CT提示双肺结构正常(图 1),术后2年肺功能检查示:用力肺活量(forced vital capacity, FVC)为2.11 L,第1秒用力呼气容积(forced expiratory volume in one second, FEV1.0)为2.04 L,FEV1.0/FVC为96.7%。
![]() 图 1 肺移植受者手术前后的胸部CT表现注:A、C图示术前肺窗;B、D图示术后2年肺窗。Figure 1. Chest CT manifestations of the lung transplant recipient before and after surgery
图 1 肺移植受者手术前后的胸部CT表现注:A、C图示术前肺窗;B、D图示术后2年肺窗。Figure 1. Chest CT manifestations of the lung transplant recipient before and after surgery2. 讨论
CF是一种侵犯多脏器的常染色体隐性遗传病,其由7号染色体上一个230 kb的基因突变引起,该基因编码含1 480个氨基酸的多肽,命名为CFTR[5]。自1989年首次发现CFTR[6],目前已知有2 000多个CFTR基因突变类型[6]。CFTR基因突变在不同人群中的频率和分布不同[7]。CFTR突变最常见于具有北欧血统的人群,其主要突变是Phe508del,约占70%[1]。亚洲人的CFTR基因突变谱可能与白种人的CFTR基因突变谱不同[8]。在2015年一项研究分析中并没有发现Phe508del在东亚血统(如中国、韩国、日本、越南和泰国)的CF患者等位基因中出现[9]。很少有突变在全球范围的流行率在0.1%以上,但一些突变在特定人群中可以达到很高的流行率[10]。例如,在丹麦82%的CF患者出现Phe508del,而在土耳其中有32%的患者出现Phe508del[11]。不同的CFTR基因突变与疾病的严重程度有关,CFTR基因功能障碍会导致一系列疾病,所涉及的器官数量和疾病严重程度各不相同[12]。
CFTR作为Cl-通道存在于上皮细胞内,同时具有Na+通道调节功能,CFTR基因突变导致外分泌腺导管上皮细胞分泌Cl-和水减少,回吸收Na+增加,外分泌液黏稠阻塞外分泌腺的管腔,细菌在黏液中繁殖,导致免疫介导的炎症反应造成多器官损害。CF患者一般幼年起病,肺部疾病的特点是反复上、下呼吸道感染和支气管扩张,常合并消化系统疾病(如胰腺充血、肠梗阻、胆汁淤积、营养不良等)以及生殖系统疾病(如先天性双侧输精管缺失、不孕不育等)[13]。其中以呼吸系统损害最为突出,呼吸系统影像学主要表现为反复气道阻塞、支气管感染引起黏液堵塞、囊性支气管扩张、支气管壁增厚[14]。这些CF患者具有发病时间早、感染程度重、病程长且反复发作、预后差、病死率高的特点,始终是临床治疗的难点。本例患者幼时起病,出现慢性鼻窦炎、反复呼吸道感染,并出现重度营养不良,内科治疗欠佳,病情迁延不愈,影像学提示双肺多发囊柱状支气管扩张伴感染,黏液栓形成,符合CF的临床及影像学表现。
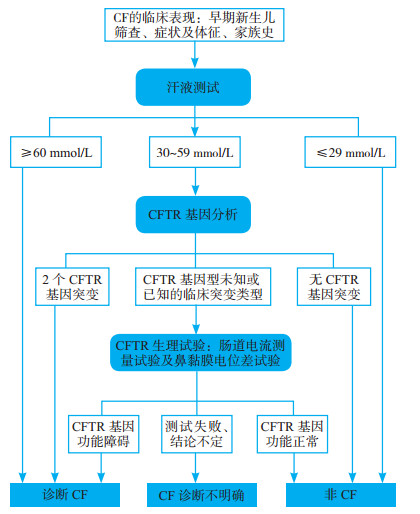
早期人们对CF的发病机制尚不清楚,治疗方法不成熟,这部分患者很少得到早期诊断。因此,CF患者幼年病死率高[15-16]。在中国,CF这类罕见病早期诊断较为困难。2016年的一项研究表明,中国大多数CF患者被误诊为支气管扩张、弥漫性细支气管炎等多种更常见的呼吸系统疾病[17]。本例患者也是早期多次被诊断为“支气管扩张”。汗液测试作为一项无创诊断手段,是诊断CF的金标准[18]。2017年CF基金会公布了CF诊断标准的最新修改版(图 2)[7]。诊断流程如下:
本例受者经过2次汗液测试,结果均高于60 mmol/L,结合受者的临床表现及体征,CF诊断明确。关于CF的治疗,在过去的60年里,通过支气管扩张剂、抗生素治疗、黏液溶解药物、胰酶替代、加强物理排痰等治疗方案和实施多学科护理,患者的生存率有了显著的提高。尽管2017年北美CF会议提出CFTR调节剂有望成为临床有效治疗CF的方法,然而也指出即使是当前最好的CFTR调节剂也只能延缓病情,仍无法阻止疾病的发展[19],任何一种药物都无法治疗存在2 000多个突变类型的疾病[20]。有研究证明可以通过基因疗法治愈CF患者,但是仍有许多突变基因类型没有明确的治疗方案[21]。肺移植是目前治疗终末期CF唯一有效的治疗方法。CF受者接受肺移植术后的近、远期生存率显著高于其他终末期肺疾病受者[22]。美国Ramos等[23]的回顾性分析提示FEV1.0/FVC < 30%是CF患者病情严重程度的重要指标,此类患者需及时转诊。因此,对于CF患者,国际心肺移植协会(International Society of Heart and Lung Transplantation,ISHLT)建议达到以下任何一条标准时应考虑肺移植:(1)CF患者FEV1.0/FVC < 30%,或尽管CF患者接受最佳治疗后FEV1.0仍在迅速下降(2)6 min步行距离 < 400 m;(3)肺动脉高压进行性加重;(4)临床病情频繁急性加重[24]。本例受者肺移植术前出现Ⅱ型呼吸衰竭、病情持续进展,并出现肺性脑病,需要长期呼吸机支持,早已到达上述各项标准,达到肺移植指征。该例CF受者接受肺移植术后经过康复治疗,肺功能逐渐恢复正常,康复后至投稿日已2年余,享有与同龄健康人同等的生活质量,提示肺移植术对CF的治疗效果非常理想。
自上世纪80年代早期首次为CF患者行肺移植以来,肺移植术取得了实质性进展,CF受者预后得到明显改善,术后5年生存率达60%~70%[25]。研究显示45%的CF受者肺移植术后生存时间超过10年[26],显著长于CF患者的中位生存时间[22]。结合此病例可见终末期CF病情进展快、感染重、内科治疗效果差,需尽快按照ISHLT建议的指征进行肺移植治疗。
综上所述,通过综合分析临床特点、影像学检查及汗液测试等手段诊断CF后,需在疾病的不同时期予以最佳治疗方式。肺移植术作为终末期CF的有效治疗手段,不仅能挽救患者生命,而且还能显著提高患者的生活质量。
-
![]()
图 1 肺移植受者手术前后的胸部CT表现
注:A、C图示术前肺窗;B、D图示术后2年肺窗。
Figure 1. Chest CT manifestations of the lung transplant recipient before and after surgery
-
[1] ELBORN JS. Cystic fibrosis[J]. Lancet, 2016, 388(10059): 2519-2531. DOI: 10.1016/S0140-6736(16)00576-6.
[2] REY MM, BONK MP, HADJILIADIS D. Cystic fibrosis: emerging understanding and therapies[J]. Annu Rev Med, 2019, 70:197-210. DOI: 10.1146/annurev-med-112717-094536.
[3] WIENCEK JR, LO SF. Advances in the diagnosis and management of cystic fibrosis in the genomic era[J]. Clin Chem, 2018, 64(6):898-908. DOI: 10.1373/clinchem. 2017.274670.
[4] ROSENBLATT RL. Lung transplantation in cystic fibrosis[J]. Respir Care, 2009, 54(6):777-787. DOI: 10.4187/002013209790983197
[5] RATJEN F, DÖRING G. Cystic fibrosis[J]. Lancet, 2003, 361(9358):681-689. DOI: 10.1016/S0140-6736(03)12567-6
[6] KEREM B, ROMMENS JM, BUCHANAN JA, et al. Identification of the cystic fibrosis gene: genetic analysis[J]. Science, 1989, 245(4922):1073-1080. DOI: 10.1126/science.2570460
[7] FARRELL PM, WHITE TB, REN CL, et al. Diagnosis of cystic fibrosis: consensus guidelines from the cystic fibrosis foundation[J]. J Pediatr, 2017, 181S:S4-S15. DOI: 10.1016/j.jpeds.2016.09.064.
[8] DE BOECK K, ZOLIN A, CUPPENS H, et al. The relative frequency of CFTR mutation classes in European patients with cystic fibrosis[J]. J Cyst Fibros, 2014, 13(4):403-409. DOI: 10.1016/j.jcf.2013.12.003.
[9] SINGH M, REBORDOSA C, BERNHOLZ J, et al. Epidemiology and genetics of cystic fibrosis in Asia: in preparation for the next-generation treatments[J]. Respirology, 2015, 20(8):1172-1181. DOI: 10.1111/resp.12656.
[10] CASTELLANI C, CUPPENS H, MACEK M JR, et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice[J]. J Cyst Fibros, 2008, 7(3):179-196. DOI: 10.1016/j.jcf.2008.03.009.
[11] MORRAL N, BERTRANPETIT J, ESTIVILL X, et al. The origin of the major cystic fibrosis mutation (delta F508) in European populations[J]. Nat Genet, 1994, 7(2):169-175. DOI: 10.1038/ng0694-169
[12] BELL SC, MALL MA, GUTIERREZ H, et al. The future of cystic fibrosis care: a global perspective[J]. Lancet Respir Med, 2020, 8(1):65-124. DOI: 10.1016/S2213-2600(19)30337-6.
[13] SANTI C, GREENE CM. Challenges facing microRNA therapeutics for cystic fibrosis lung disease[J]. Epigenomics, 2020, 12(3): 179-181. DOI: 10.2217/epi-2019-0395.
[14] DAVIS PB. Cystic fibrosis since 1938[J]. Am J Respir Crit Care Med, 2006, 173(5):475-482. DOI: 10.1164/rccm.200505-840OE
[15] MURPHY KP, MAHER MM, O'CONNOR OJ. Imaging of cystic fibrosis and pediatric bronchiectasis[J]. AJR Am J Roentgenol, 2016, 206(3):448-454. DOI: 10.2214/AJR.15.14437.
[16] LEWIS PA, MORISON S, DODGE JA, et al. Survival estimates for adults with cystic fibrosis born in the United Kingdom between 1947 and 1967. the UK CysticFibrosis Survey Management Committee[J]. Thorax, 1999, 54(5):420-422. DOI: 10.1136/thx.54.5.420
[17] SHEN Y, LIU J, ZHONG L, et al. Clinical phenotypes and genotypic spectrum of cystic fibrosis in Chinese children[J]. J Pediatr, 2016, 171:269-276. DOI: 10.1016/ j.jpeds.2015.12.025.
[18] SAVANT AP, MCCOLLEY SA. Cystic fibrosis year in review 2018, part 1[J]. Pediatr Pulmonol, 2019, 54(8):1117- 1128. DOI: 10.1002/ppul.24361.
[19] MARTINIANO SL, TOPRAK D, ONG T, et al. Highlights from the 2017 North American Cystic Fibrosis Conference[J]. Pediatr Pulmonol, 2018, 53(7):979-986. DOI: 10.1002/ppul.24000.
[20] RAFEEQ MM, MURAD HAS. Cystic fibrosis: current therapeutic targets and future approaches[J]. J Transl Med, 2017, 15(1):84. DOI: 10.1186/s12967-017-1193-9.
[21] STRUG LJ, STEPHENSON AL, PANJWANI N, et al. Recent advances in developing therapeutics for cystic fibrosis[J]. Hum Mol Genet, 2018, 27(R2):R173-R186. DOI: 10.1093/hmg/ddy188.
[22] CHAMBERS DC, CHERIKH WS, GOLDFARB SB, et al. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: thirty-fifth adult lung and heartlung transplant report-2018; focus theme: multiorgantransplantation[J]. J Heart Lung Transplant, 2018, 37(10):1169-1183. DOI: 10.1016/j.healun.2018.07.020.
[23] RAMOS KJ, QUON BS, HELTSHE SL, et al. Heterogeneity in survival in adult patients with cystic fibrosis with FEV1 < 30% of predicted in the United States[J]. Chest, 2017, 151(6):1320-1328. DOI: 10.1016/j.chest.2017.01.019.
[24] WEILL D, BENDEN C, CORRIS PA, et al. A consensus document for the selection of lung transplant candidates: 2014--an update from the Pulmonary Transplantation Council of the International Society for Heart and Lung Transplantation[J]. J Heart Lung Transplant, 2015, 34(1):1-15. DOI: 10.1016/j.healun.2014.06.014.
[25] HIRCHE TO, KNOOP C, HEBESTREIT H, et al. Practical guidelines: lung transplantation in patients with cystic fibrosis[J]. Pulm Med, 2014:621342. DOI: 10.1155/2014/621342.
[26] SNELL G, REED A, STERN M, et al. The evolution of lung transplantation for cystic fibrosis: a 2017 update[J]. J Cyst Fibros, 2017, 16(5):553-564. DOI: 10.1016/j.jcf. 2017.06.008.
-
期刊类型引用(2)
1. 王梓竹,王昊,陈兰勤,贺建新,徐保平,申昆玲. 肺移植治疗儿童囊性纤维化1例. 中华实用儿科临床杂志. 2022(03): 216-218 .  百度学术
百度学术
2. 王胜飞,陈静瑜,刘懿,郑明峰,毛文君. 双肺移植治疗儿童囊性纤维化的多学科综合诊疗. 器官移植. 2021(02): 184-190 . 本站查看
其他类型引用(1)
 下载:
下载:
计量
- 文章访问数: 267
- HTML全文浏览量: 125
- PDF下载量: 22
- 被引次数: 3


